We use mathematics to unravel the invisible in biology
MACHINE LEARNING AND AI METHODS FOR SYSTEMS BIOLOGY
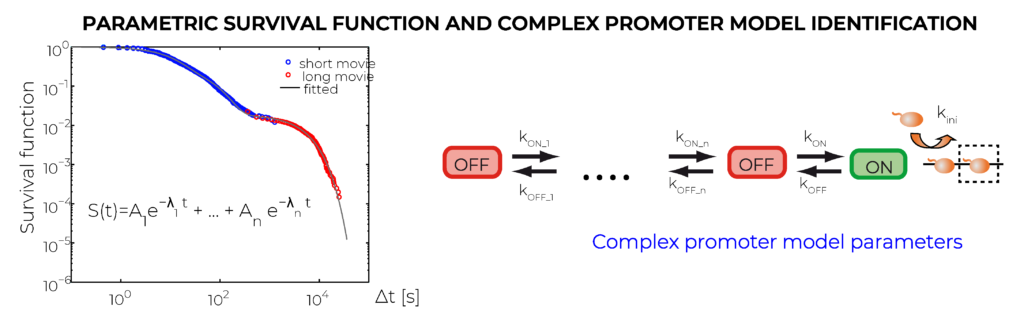
Hybrid symbolic/numeric method for reverse engineering of transcriptional bursting processes
We use machine learning to derive characteristics of single cell transcription activity from real time mRNA pre-synthesis data. The nascent mRNA generated by an active transcription site is identified via the interaction between MS2 proteins fused to a fluorescent protein and specific hairpins purposely inserted in the sequence. The MS2 signal is the convolution of the signal from a polymerase and the time sequence of transcription events. Using a deconvolution method and high resolution movies, we generate a time map of transcription events indicating, for each cell, the moments when different RNAP molecules start producing mRNA. The map can be used for direct characterisation of transcription features, such as polymerase convoys and various statistics of inter-event times. Another output of the approach is a multiscale cumulative distribution function of the waiting time separating succesive transcription events (or the complementary function, called survival function). This distribution, obtained by combining short, high-resolution and long, low-resolution movies, covers timescales from second to 10 hours. The last output of our method is the transcription model identification. To this end, we use exact symbolic solutions, relating the parameters of the multiscale distributions to kinetic parameters of a model.
References
K. Tantale, E. Garcia-Oliver, A. L’Hostis, Y. Yang, MC. Robert, T. Gostan, M. Basu, A. Kozulic-Pirhern, JC. Andrau, F. Muller, E. Basyuk*, O. Radulescu*, E. Bertrand*. Stochastic pausing at latent HIV-1 promoters generates transcriptional bursting. 2020, in revision Nature Communications. *corresponding authors. Bioarxiv doi: https://doi.org/10.1101/2020.08.25.265413.
V.L. Pimmet, M.Dejean, C.Fernandez, A.Trullo, E.Bertrand, O.Radulescu, M.Lagha. Quantitative imaging of transcription in living Drosophila embryos reveals the impact of core promoter motifs on promoter state dynamics. 2020, in revision Nature Communications. Bioarxiv doi:
https://doi.org/10.1101/2021.01.22.427786.